ASE 使用
# ASE 使用
# 介绍
atomic simulation environment (ASE) (opens new window):一系列用于设置、操作、运行、可视化及分析原子模拟的工具和 Python 模块
ASE 通过
Calculators为不同的计算代码(DFT/MD)提供接口,Calculators与核心Atomsobject 和 ASE 中的许多可用算法一起使用。

注:ase 网站中的代码源码参数及注释与安装的 python package 源码会有不一致的地方,写脚本还是以 pacakge 的源码为准
from ase.atoms import Atoms
from ase.calculators.singlepoint import SinglePointCalculator
results={"energy": -7.0}
atoms.calc = SinglePointCalculator(atoms, **results)
atoms.get_potential_energy()
# ase rdf 计算
from ase.geometry.analysis import Analysis
# 添加 element 参数,可计算 partial rdf;默认计算 full rdf
rdf = Analysis(images=...).get_rdf()
ase 缺陷计算 寻找最优的超胞形状
Tools for defect calculations — ASE documentation (opens new window)
# 添加真空层;单独使用该函数时,返回值为 None,即无效果
from ase.build import add_vacuum
# 优化器
from ase.optimize.lbfgs import LBFGS
from ase.optimize import QuasiNewton
from ase.constraints import FixAtoms
# 振动分析
from ase.vibrations import Vibrations
from ase.data import atomic_numbers
from ase.md.verlet import VelocityVerlet
from ase.units import fs
# 获取布拉维点阵
atoms.cell.get_bravais_lattice()
# 简易元素周期表绘制
# reference: https://wiki.fysik.dtu.dk/ase/gallery/gallery.html
from ase.utils.ptable import ptable
atoms = ptable()
atoms.write("ptable.png")
interface 构建(较简单情况):Interface building - Manipulating atoms — ASE documentation (opens new window)
DOS、能带、EOS 计算:Crystals and band structure — ASE documentation (opens new window)
# 参考资料
ase 教程(内容较详细)
ase 进行 md 计算 package
弹性张量相关,ASE 接口:GitHub - jochym/Elastic: A module for ASE for elastic constants calculation. (opens new window)
ase tutorial
基于 PAW 和 ASE 的 DFT code
GPAW: DFT and beyond within the projector-augmented wave method — GPAW (opens new window)
ase 相关脚本案例
GitHub - jkitchin/dft-book: A book on modeling materials using VASP, ase and vasp (opens new window)
ase 结构 2D 和 3D 渲染
GitHub - superstar54/x3dase: X3D for Atomic Simulation Environment (opens new window)
ase.lattice 有生成 graphene 和 graphite modules
ase/lattice/hexagonal.py · master · ase / ase · GitLab (opens new window)
ase symmetry 教程(内容一般)
表面吸附、EOS、弹性常数计算(ASE 中无计算弹性常数的模块和类)
GitHub - jochym/Elastic: A module for ASE for elastic constants calculation. (opens new window)
Calculation of elastic properties of crystals — Elastic v5.1.0 documentation (opens new window)
# 安装
pip install ase # 安装
ase test # 测试;需安装 pytest
# 常用模块
from ase.cell import Cell
# cell 参数转换成 cell matrix
cell = Cell.fromcellpar([3.31, 3.31, 3.31, 90, 90, 90])
cell[:]
构型可视化
# 方式 1
from ase.visualize.plot import plot_atoms
plot_atoms(atoms)
# 方式 2
from ase.visualize import view
view(atoms, viewer="ngl")
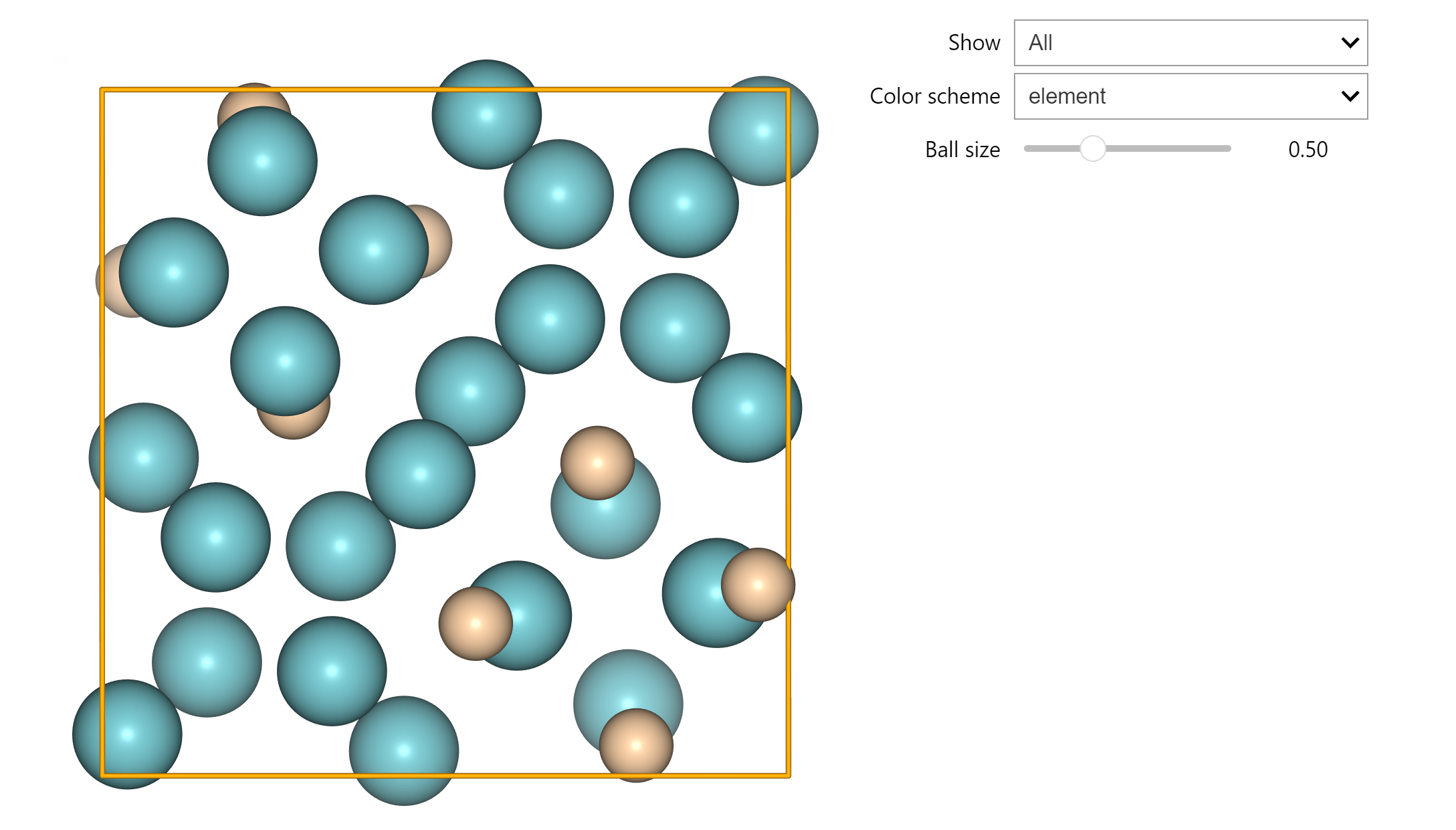
nglview,可在 jupyter notebook 中可视化构型
pip install nglview
nglview 效果图:
crystal 构建
# 方式 1;最简单
from ase.build import bulk
# 方式 2
from ase.atoms import Atoms
# 方式 3
from ase.spacegroup import crystal
from ase.spacegroup import Spacegroup
spg = Spacegroup(152)
# 查看等同原子坐标
spg.equivalent_sites([0.4673, 0, 0.3333])
超胞
# 方式 1
supercell = atoms * (2, 2, 2)
db.select(sort) 中的 sort 为 含 key 的 str,含 - 时,降序
ase neb 方法:Nudged elastic band — ASE documentation (opens new window)
ase 相图绘制(2 维,3 维):Phase diagrams and Pourbaix diagrams — ASE documentation (opens new window)
from ase.build import sort
# 按照 chemical symbols 排序生成新的 Atoms object
sort(atoms)
# 固定原子
from ase.constraints import FixAtoms
atoms = ...
# 按照原子类型或 z 轴坐标进行固定
c = FixAtoms(mask=atoms.symbols == 'Cu')
c = FixAtoms(mask=atoms.positions[:, 2] < 1.0)
atoms.set_constraint(c)
Atomic Simulation Environment (ASE) 基础用法 (opens new window)
# 执行 eos 计算,拟合体模量 B
from ase.eos import calculate_eos
from ase.units import kJ
from ase.atoms import Atoms
calc = ...
atoms: Atoms = ...
atoms.calc = calc
eos = calculate_eos(atoms, trajectory="XXX.traj")
v, e, B = eos.fit()
print(B / kJ * 1.0e24, "GPa")
# CLI
开启 ase 补全(适用 bash,zsh 不行)
ase completion >> ~/.bashrc
# 列出 ase 可识别的构型文件格式
ase info --formats
# 列出 ase 的 calculators 以及是否被安装
# 3.22.1 与 3.23.0 版本的输出格式有区别
ase info --calculators
# 构型转换
ase convert -i vasp -o xyz -f -v POSCAR structure.xyz
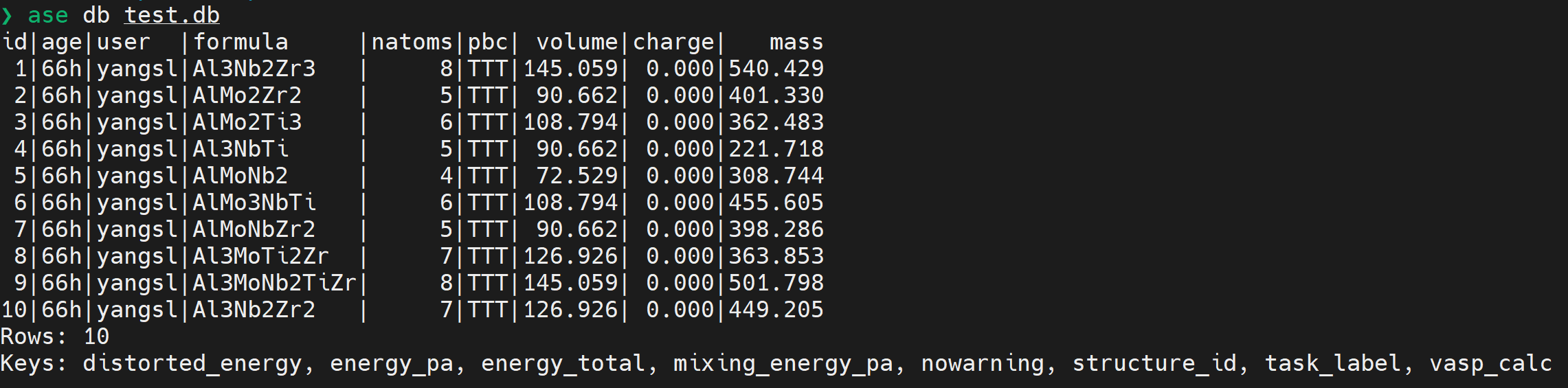
# 查看 db 文件内容 推荐
ase db test.db
-L N # 只显示前 N 行
--offset N # 跳过前 N 行
--show-keys # 显示所有 keys
--show-values key1,key2,... # 显示 key 的值;value为数值时,只显示首尾值,如 energy_pa: [-9.18438289..-5.855563642]
# ase.atoms
array methods of Atoms objects (opens new window)
Atom 和 Atoms 是 ASE 的两个基本 Object, Atoms 由 Atom 构成。 本质上 Atoms 是 Atom 的 list,可以使用标序的方式来查看 Atom
需添加 calculator 才能使用的 methods:Adding a calculator (opens new window)
get_potential_energy()
get_forces()
get_stress()
- 属性 -
- 方法
get_XXX()method -set_XXX()method -
from ase.atoms import Atoms
from ase.formula import Formula
atoms: Atoms
# 常用属性和方法
# 化学式
formula = atoms.get_chemical_formula()
# 成分 {'Al': 5, 'Ti': 1}
composition = Formula(formula).count()
# 原子数
natoms = len(atoms)
# 元素种类数
nele = len(set(atoms.get_chemical_symbols()))
# 删除 H 原子
del atoms[[atom.index for atom in atoms if atom.symbol == "H"]]
# 生成 slab 模型;在指定轴两端各添加真空层 vacuum 数值并使原子位点居中
center(vacuum=10.0, axis=2)
copy() # 拷贝
# 已施加 PBC 时,可将胞外原子移至胞内;下面的 wrap 参数同
wrap()
write() # 写入构型格式文件
# 分数坐标;wrap 参数默认为 True
get_scaled_positions()
# 笛卡尔坐标;wrap 参数默认为 False
get_positions()
positions
get_distance() # 两原子间的距离
get_distances() # 第 i 个原子与给定原子列表间的距离
# ase.build
# bulk
简单 bulk 模型构建 示例代码
from ase.build import bulk
# 原胞
primCell = bulk("Al", "fcc", a=4.05)
# 单胞 cubic=True
unitCell = bulk("Al", "fcc", a=4.05, cubic=True)
# 超胞
superCell = unitCell * (2, 2, 2)
# surface
from ase.build import surface
简单 bulk 模型的表面构建 示例代码
# ase.io
- 构型格式文件读入、写出
- 函数
read()可自动识别文件格式;ase 中可识别的文件格式(部分格式只有read或write一个函数):File input and output — ASE documentation (opens new window) - 可以读取 gz 格式压缩文件,如 OUTCAR.gz
lammps的结构文件转其他格式 - 知乎 (opens new window)
写法一:在 write() 函数中的 format 参数指定文件格式
from ase.io import read, write
write(filename=..., images=..., format=...)
写法二:从 ase.io 中导入具体格式的模块及其函数
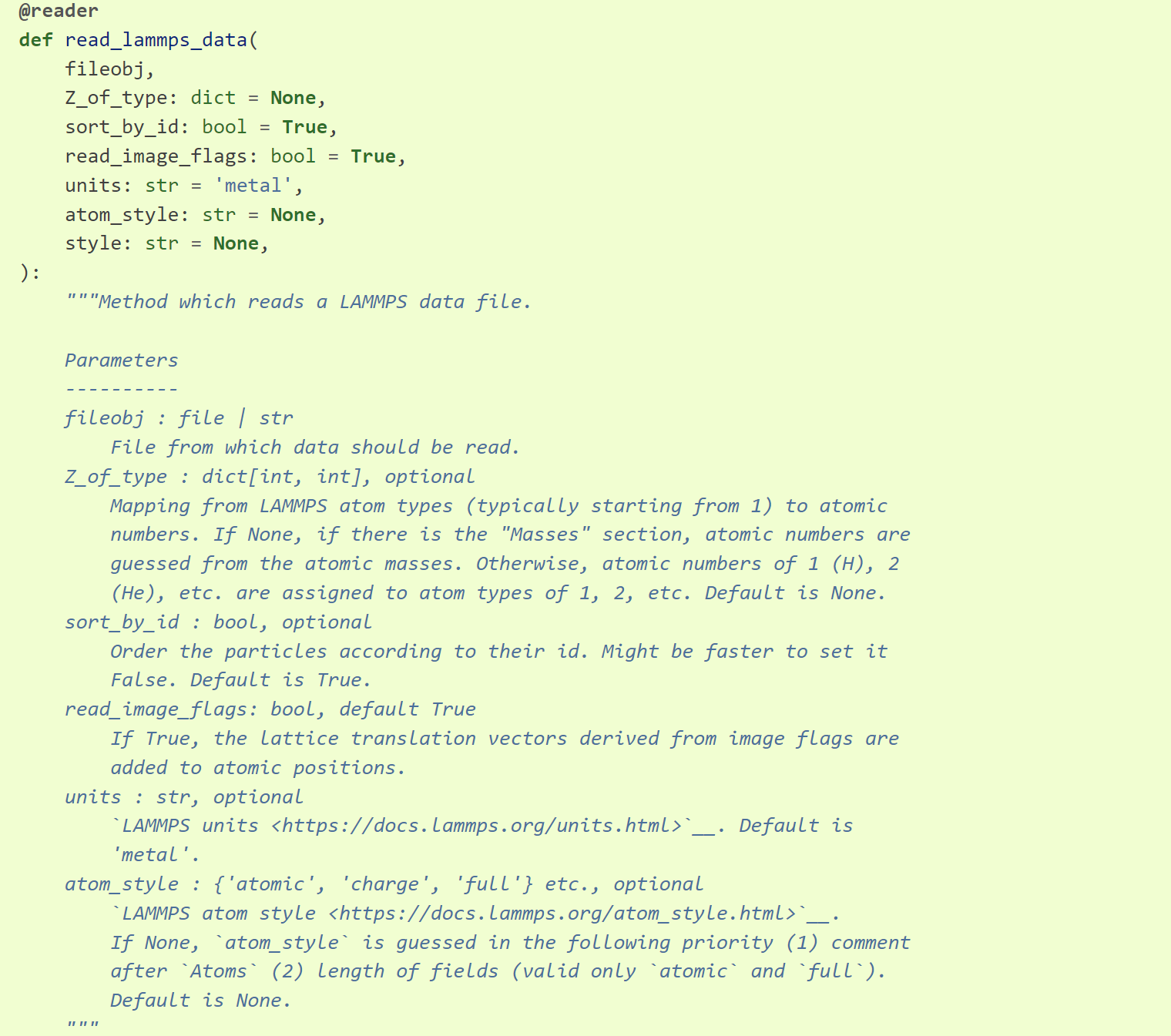
# LAMMPS data 格式
from ase.io.lammpsdata import read_lammps_data, write_lammps_data
# vasp 格式
from ase.io.vasp import read_vasp, write_vasp
# VASP 输出文件格式
from ase.io.vasp import read_vasp_out
# material studio xsd 格式
from ase.io.xsd import read_xsd, write_xsd
extxyz.py 源代码相关 warning:
/home/yangsl/src/miniconda3/envs/base_ysl/lib/python3.11/site-packages/ase/io/extxyz.py:1000: UserWarning: write_xyz() overwriting array "forces" present in atoms.arrays with stored results from calculator
warnings.warn('write_xyz() overwriting array "{0}" present '
# ase.eos
获取平衡体积,能量和体模量
from ase.eos import EquationOfState
from ase.units import kJ
# murnaghan birch vinet
eos = EquationOfState(volumes, energies, eos="birchmurnaghan")
v0, e0, B = eos.fit()
print(f"v0 = {v0:.3f}")
print(f"e0 = {e0:.3f}")
print(f"B = {B / kJ * 1.0e24:.1f} GPa")
ax = eos.plot()
ax.set_title(label=None)
# ase.db
from ase.db import connect
from ase.db.row import AtomsRow
db_fn = ...
db = connect(db_fn)
# 给 db 添加元数据
db.metadata = {...}
# 获取 db 文件中存储的结构数目
len(db)
db.count()
# 添加 selection 筛选条件
db.count("vasp_calc=Yes")
# 筛选 id<=5 的所有结构
# selection 可以是 id 或其他 AtomsRow 中的 key
# 注:字符与符号之间不能有空格
for row in db.select("id<=5"):
...
# 筛选 id>=5, id<=10 的所有结构
for row in db.select("id>=5, id<=10"):
...
# 单个 AtomsRow
# id 从 1 开始
row = db.get(id=10)
# 获取 AtomsRow 的 keys
print(row._keys)
# 获取 AtomsRow 的 key_value_pairs
print(row.key_value_pairs)
# 根据 key 获取 value
print(row.vasp_calc)
# 单个 Atoms
atoms_specific = db.get_atoms(id=10)
# 将 db 中的 AtomsRow 的结构和数据写入到其他 db 文件
db_output_fn = "..."
db_output = connect(db_output_fn)
for row in db.select("id<=10"):
key_value_pairs = row.key_value_pairs
data = row.data
# 将 AtomsRow 转化成 Atoms
atoms = row.toatoms()
db_output.write(atoms=atoms, key_value_pairs=key_value_pairs, data=data)
# ase.calculators
effective medium theory (EMT)
from gpaw import GPAW
from ase.calculators.emt import EMT
atoms = ...
calc = GPAW(mode='lcao', basis='dzp', txt='gpaw.txt')
atoms.calc = calc
opt = BFGS(atoms, trajectory='opt.traj')
opt.run(fmax=0.05)
# VASP
设置 VASP 执行命令和赝势路径
export ASE_VASP_COMMAND="mpirun path/vasp_std"
export VASP_PP_PATH=path/pp_path
或者在脚本中添加如下命令以设置相关环境变量
import os
os.environ["ASE_VASP_COMMAND"] = ...
os.environ["VASP_PP_PATH"] = ...
ASE VASP Calculator 赝势不同泛涵目录命名
LDA: $VASP_PP_PATH/potpaw/
PBE: $VASP_PP_PATH/potpaw_PBE/
PW91: $VASP_PP_PATH/potpaw_GGA/
# 查找元素 POTCAR 示例:potpaw_PBE/N/POTCAR
from ase.calculators.vasp import Vasp
calc = Vasp(
istart=0,
icharg=2,
encut=400,
ismear=1,
sigma=0.2,
lreal="Auto",
kpts=[5, 5, 5],
ivdw=12,
ediff=1e-05,
lwave=False,
lcharg=False,
)
# LAMMPS
from ase.calculators.lammpsrun import LAMMPS
parameters = {
"pair_style": "meam/c",
"pair_coeff": ["* * library.meam Au Au.meam Au"],
}
files = ["library.meam", "Au.meam"]
calc = LAMMPS(parameters=parameters, files=files)
在 Pi 中用 ASE 的 VASP 的 Calculator
申请节点运算(不写提交脚本)
srun -p small -n 4 --pty /bin/bash
~/.bashrc文件添加内容
module purge
module load intel-oneapi-compilers/2021.4.0
module load intel-oneapi-mpi/2021.4.0
module load intel-oneapi-mkl/2021.4.0
export I_MPI_PMI_LIBRARY=/usr/lib64/libpmi.so
export I_MPI_FABRICS=shm:ofi
export ASE_VASP_COMMAND="srun --mpi=pmi2 /lustre/home/acct-mseklt/mseklt/yangsl/bin/vasp_std"
python *.py
写提交脚本
~/.bashrc文件添加内容
export ASE_VASP_COMMAND="srun --mpi=pmi2 /lustre/home/acct-mseklt/mseklt/yangsl/bin/vasp_std"
submit_ase.slurm脚本
#!/bin/bash
#SBATCH -J vasp
#SBATCH -p small
#SBATCH -N 1
#SBATCH --ntasks-per-node=4
#SBATCH -o %j.out
#SBATCH -e %j.err
module purge
module load intel-oneapi-compilers/2021.4.0
module load intel-oneapi-mpi/2021.4.0
module load intel-oneapi-mkl/2021.4.0
export I_MPI_PMI_LIBRARY=/usr/lib64/libpmi.so
export I_MPI_FABRICS=shm:ofi
python python-file-name.py
sbatch submit_ase.slurm
在思源中用 ASE 的 VASP 的 Calculator
- 提交脚本(
ase-submit-sy.slurm)
#!/bin/bash
#SBATCH -J ase
#SBATCH -p 64c512g
#SBATCH -N 1
#SBATCH --ntasks-per-node=2
#SBATCH --exclusive
#SBATCH -o %j.out
#SBATCH -e %j.err
module load vasp/5.4.4-intel-2021.4.0
ulimit -s unlimited
python python-file-name.py